ABSTRACT
OBJECTIVES: present a case of Inborn errors of immunity (IEI) as a potential diagnosis in pediatric patients with recurrent infections.
DESCRIPTION: male patient, 13 years old, since he was eight months old had recurrent diarrhea, sinusitis, otitis, abscesses and urinary tract infections. At the age of ten, he presented mastoiditis progressing to meningitis, he was admitted to a tertiary hospital, where an immunological evaluation was performed, which led to the diagnosis of Predominantly Antibody Deficiency (PAD), with suspected X-linked Agammaglobulinemia (XLA). Treatment was initiated with administration of intravenous gamma globulin 400 mg/kg every four weeks, with a significant improvement of the condition.
DISCUSSION: usually, the diagnosis of XLA tends to be made in the first three years of life. However, in this report, although the first manifestations started at eight months of age, there was a delay of ten years before starting the treatment. In fact, the diagnosis of children and adults with IEI can be delayed if healthcare professionals are unable to find the true cause of recurrent infections. Therefore, the relevance of considering such pathologies in the presence of risk signs is highlighted, as early diagnosis being essential in treating and preventing morbidities.
Keywords:
Primary immunodeficiency diseases, Immunity, Infections
RESUMO
OBJETIVOS: apresentar um caso de Erro Inato da Imunidade (EII) como diagnóstico em potencial de pacientes pediátricos com infecções de repetição.
DESCRIÇÃO: paciente masculino, 13 anos, desde os oito meses de idade apresentou quadros repetidos de diarreias, sinusites, otites, abscessos e infecções do trato urinário; destacando-se a otite, sinusite e diarreia pela maior recorrência. Aos dez anos, quando apresentou mastoidite evoluindo para meningite, foi internado em um hospital terciário, onde foi realizada avaliação imunológica, a qual levou ao diagnóstico de Deficiência Predominantemente de Anticorpos (DPAs), tendo como suspeita a agamaglobulinemia ligada ao cromossomo X (ALX). Foi iniciado tratamento com administração de gamaglobulina endovenosa 400 mg/kg a cada quatro semanas, ocorrendo melhora significativa do quadro.
DISCUSSÃO: normalmente, o diagnóstico da ALX tende a ser feito nos primeiros três anos de vida. Neste relato, entretanto, embora as primeiras manifestações tenham iniciado aos oito meses de idade, ocorreu um atraso de dez anos até o início do tratamento. De fato, o diagnóstico de crianças e adultos com EII pode ser retardado se os profissionais de saúde não conseguirem encontrar a causa das infecções recorrentes. Destaca-se, portanto, a relevância de se considerar tais patologias na vigência de sinais de riscos, pois o diagnóstico precoce é fundamental para tratar e prevenir morbidades.
Palavras-chave:
Doenças da imunodeficiência primária, Imunidade, Infecções
IntroductionInborn errors of immunity (IEI) constitute of a group with more than 430 diseases that can cause various manifestations in the immune system functions. Among this universe of disorders that constitute of IEI, predominantly antibody deficiencies (PAD) and immunodeficiency combined are the most frequent, and male individuals with family consanguinity are more affected. These pathologies result in genetic defects, and their expression usually occurs at childhood, through repeated infections, such as otitis, sinusitis, stomatitis, and pneumonia. Early diagnosis is essential to institute treatment, as well as to define the best management for the IEI type, thus preventing the morbidity that is linked to the condition.
1-3X-linked agammaglobulinemia (XLA) is a classification of IEI in which antibody deficiency is predominant. XLA consists of absent or decreased B lymphocytes and immunoglobulins. Affected individuals are susceptible to bacterial infections by encapsulated agents and to enteroviruses. In 85% of the cases, its origin is linked to an error on the X chromosome, generating defects in the Bruton tyrosine kinase (BTK) gene, and for this reason it is called X-linked agammaglobulinemia. The error in this gene causes reduced or absent serum levels of IgM, IgG, IgA, IgE and B lymphocytes, as well as reduced size of the tonsils, spleen, adenoids and Peyer's patches. This is because BTK is expressed on all hematopoietic cell lines, with the exception of the T cells and plasma cells.
4In the physical examination of patients with XLA and some IEI, it is common for the tonsils and cervical lymph nodes to be small or even absent.
1 In addition, because of recurrent otitis, otoscopy often shows lesions or perforations of the tympanic membranes. As a result of upper and lower airway infections, complaints of chronic cough and purulent secretion are frequent. Crepitant lung rales are recurrent during pulmonary auscultation.
DescriptionA 13-year-old mixed colored skin male patient from the city of Patos, Paraíba. At eight months of age, he started with a severe diarrhea, requiring hospitalization, and since then he has had recurrent diarrhea. In the first year of life he started to present sinusitis, which became recurrent. At the age of two and three, he developed an abscess on the abdominal wall and a facial abscess, requiring hospitalization. At seven, he was hospitalized for hematuria and urinary tract infection. After that, he was stricken with otitis, whose cultures were positive for
Candida sp. and
Staphylococcus aureus, with an interval of five months between the situation presented.
At the age of ten, he presented mastoiditis evolving into meningitis, and was admitted to the University Hospital in the state capital, and the immunological evaluation was extended and the results of which are described in Table 1. The exams showed decreased immunoglobulins, absence of B lymphocytes, normal T lymphocytes, and NK cells with reduced levels, which led to the syndromic diagnosis of predominantly antibody deficiency (PAD), and administration of intravenous gamma globulin (IVG) of 400 mg/kg was initiated every four weeks. As mentioned above, B lymphocytes, antibody-producing cells, are absent or reduced in the presence of this syndrome, while T lymphocytes, in general, do not alterate, because they are part of the cellular immunity.
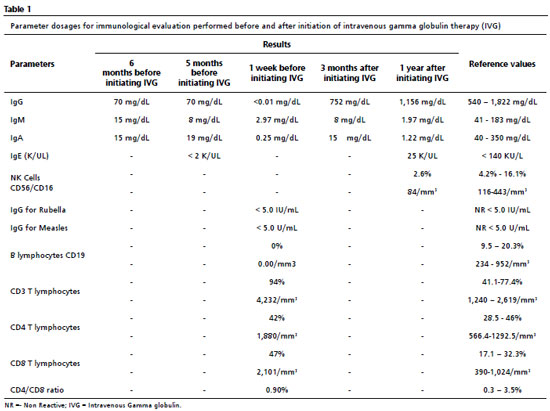
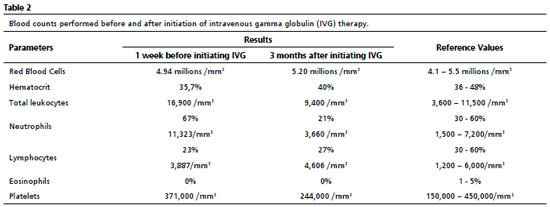
Thereafter, there was a significant improvement in the clinical and laboratorial findings, as shown in Tables 1 and 2. After initating continuous treatment with a monthly of IVG replacement, the patient presented only one episode of urinary tract infection, and the urine culture showed Escherichia coli, using ceftriaxone 30 mg/kg at home treatment. From the first manifestation of immunodeficiency until the appropriate diagnosis, ten years had passed.During this time, the patient missed one year of school due to school absences, which may also be related to the moderate degree of mixed hearing loss in the right ear, revealed by the audiological evaluation.
Thinking about other causes of immunodeficiencies, a serological test for Human Immunodeficiency Virus (HIV) was requested, the result of which was "non-reactive". For etiological confirmation of the PAD, a genetic test was requested, which confirmed the main hypothesis of X-linked agammaglobulinemia, detected by sequencing of the BTK exome in hemizygosis.
As for personal history, the mother reported prenatal care, and the patient was born at 40 weeks of gestational age and weighed 3,435 kg. The fall of the umbilical stump occurred on the eighth day. He received all the vaccines from the
Programa Nacional de Imunização (National Immunization Program) and no adverse reaction was reported. Exclusive breastfeeding lasted until three months of age.
As for autoimmune and allergic diseases and other comorbidities, only vitiligo was reported. He was hospitalized seven times in all, with a maximum of 22 days in one hospitalization. Intensive care was required during hospitalization for meningitis.
There is no consanguinity in the family history, however the death of his mother's brother at the age of 16 from recurrent infections contributed to the diagnosis.
DiscussionAs mentioned above, the clinical manifestations of IEI begin during childhood, although in some cases they may occur after the second or third decade of life. The mentioned patient presented his first manifestations quite early, at eight months of age. In general, the diagnosis of congenital agammaglobulinemia (CA) tends to be made most commonly in the first three years of life.
5 However, in this report, the boy went through a debilitating ten-year path to a suspicious diagnosis. Indeed, the diagnosis of children and adults with IEI can be delayed if health care professionals cannot find the true cause of the recurrent infections. This has been reported in other cases in the literature.
5,6The spectrum of infections in IEI is one of the main factors for the suspicion of the diagnosis in children during their early years of life, as well as being a parameter for follow-up during the treatment.
4 In this sense, the male patient may have the diagnostic hypothesis for XLA considering when there are recurrent infections during early childhood, a pathological family history positive for PADs, the occurrence of hospitalization before the age of five, and vaccine reaction, especially after vaccination with live attenuated virus. The patient had all these conditions, except for the vaccine reactions. The
Sociedade Brasileira de Imunizações (SBIm) (Brazilian Society of Immunizations) has available in its arsenal for children from zero to five years of age the following live attenuated virus vaccines: Oral Poliomyelitis, Monovalent Rotavirus, Pentavalent Rotavirus, Yellow Fever, MMR (measles, mumps, and rubella), Varicella, and Dengue.
7 These vaccines should be avoided in patients with suspected or diagnosed for XLA.
The laboratorial tests performed to initiate the investigation of CA aimed to confirm the existence of the immunological defect, thus, they must contain blood count, dosage of immunoglobulins (IgM, IgG, IgA and IgE) and titling of antibodies generated in response to the vaccines belonging to the vaccination framework in force. The second stage of testing should contain a cytometry flow in order to phenotype and count the lymphocytes.In the present case, after immunological evaluation, a reduction in serum levels of immunoglobulins and B lymphocytes was noted, as well as the absence of antibodies against rubella and measles, and the presence of normal serum levels of T-lymphocytes. Moreover, the confirmation of the diagnosis can be made by genetic testing that confirms variants in the BTK gene, or, if genetic testing is not available, a confirmed family history of XLA.
4 Since it was not possible to confirm the family history, a genetic test should be requested, which identifies, the hemizygosis, in the BTK gene, the variant ChrX:101.356.055 G>T, promoting the substitution of the aspartate amino acid in codon 521 by glutamate (p.Asp521Glu).Based on this confirmation of X-linked agammaglobulinemia, the patient was classified according to the International Classification of Diseases (ICD-10) as D80.0.
Aspartate at position 521 is highly conserved in diverse biological species, and "in silico" pathogenicity prediction computer programs suggest that its replacement by glutamate is potentially deleterious. Approximately this variant is absent among 181,000 X chromosomes in population-based individuals and has been previously described in the medical literature in association with the agammaglobulinemia condition.
8,9 It is worth noting that genetic testing is not essential to initiate the treatment in being suspected of IEI. In the present case, such a test was performed only about two years after initiating therapy.
From the syndromic diagnosis of PADs, several etiologies can be considered. Among patients with circulating B cells below 2% of the total of lymphocytes and with undetectable or very low antibody levels, about 85 to 90% have XLA, due to mutations in the BTK gene, and 5% of them have immunoglobulin M heavy chain deficiency, a condition whose clinical manifestations are more severe compared to XLA. Patients in this case are at greater risk for pseudomonas sepsis, arthritis, skin abscesses, chronic diarrhea, and enteroviral infections in the central nervous system (CNS).
10,11In the common variable immunodeficiency (CVID), there is reduced serum levels of at least two immunoglobulin isotypes, associated with normal or reduced amounts of B cells. It is usually diagnosed between the third and the fifth decade of life, and when it begins in childhood, it is usually associated with medium otitis, growth deficit, and developmental delay.
10Treatment for IEI depends on the disease diagnosed and can be definitive or supportive. The therapeutic options are still under study and aim to prevent and treat infections, stimulating the immune system, and treating the cause of immunodeficiency. Given the diagnostic hypothesis, human immunoglobulin replacement is the main therapeutic method in patients with IEI in almost 75% of this disease group.
12 Administration can be done intravenously (IVG) or subcutaneously (SCIg), but in urgent cases, IVG is the route used. Intravenous immunoglobulin replacement has a dosage range from 400 mg/kg/dose to 800 mg/kg/dose by administrating every 21 to 28 days. The dose and interval can be adjusted to decrease the incidence of infection. For subcutaneous replacement, it is recommended that therapy should be initiated using 100 to 200 mg/kg/dose every seven days.
12,13 Immunoglobulin treatment should be continuous.
After initiation of immunoglobulin therapy, it is expected that severe infections, such as sepsis, meningitis, and cellulitis would be reduced; however, it is common for patients to continue to have chronic and/or less severe infections, such as sinusitis, urinary tract infections, and otitis.
2 Since the suspected diagnosis of XLA, the mentioned patient initiated immunoglobulin replacement with intravenous administration of IgG every four weeks, achieving considerable improvement of the symptoms.
Prophylaxis antibiotic is another therapeutic method that aims to reduce the risk of complications and is patient-specific.In the case in question, it has not been necessary to use this resource so far.
An alternative therapy for IEI is hematopoietic stem cell transplantation (HSCT), known as bone marrow transplant, which consists of replacing altered cells with hematopoietic stem cells from healthy donors.It is a curative treatment option for specific cases of IEI, but its indication should be individualized and should consider the risks of future disease progression versus the risks of transplantation, such as graft versus host disease.In general, HSCT is not indicated for patients with XLA.
14Gene therapy, finally, constitutes a promising possibility of cure for patients with IEI, since it corrects the genetic alterations of the individual´s hematopoietic stem cells, restoring B-lymphocyte function, without the need to wait for compatible donors and with less risk of rejection.
14Based on the discussion of this case report, it can be stated that the diagnosis of IEI is still a challenge for health professionals, as many cases take years to receive the appropriate treatment. Therefore, it is recommended that future research study the main factors that prevent the early diagnosis of such diseases.Concomitantly, it is of fundamental importance that health education work be carried out in this sense, in order to train more and more professionals and, consequently, avoid complications resulting from the disease, reduce hospitalizations and health expenses, as well as promote a better quality of life for affected individuals.
References1. Rodrigues LS, Vernizzi CC, Ikuno CM, Menezes CT, Silva CM, Carvalho LR,
et al. Considerações sobre erros inatos da imunidade - um desafio diagnóstico na pediatria. In: FF Carvalho Junior. Alergia e imunologia: abordagens clínicas e prevenções. São Paulo: Científica Digital. 2021. p.55-77.
2. Ferreira JFS. Imunodeficiências primárias na infância - quando o pediatra deve suspeitar e como deve se conduzir? Rev Saúde Criança Adolesc. 2011; 3 (1): 58-62.
3. Pinto-Mariz F. Failure of immunological competence: when to suspect? J Pediatr (Rio J). 2021; 97 (Supl. 1): S34-8.
4. Vilela MM. Human Inborn Errors of Immunity (HIEI): predominatly antibody deficiencies (PADs): if you suspect it, you can detect it. J Pediatr (Rio J). 2021; 97 (Supl. 1): S67-74.
5. Estrella L, Foley ME, Cunningham-Rundles C. X-linked agammaglobulinemia in a 10-year-old child: A case study. J Am Acad Nurse Pract. 2007 Apr; 19 (4): 205-11.
6. Botek M, Krejčí J, Neuls F, Novotný J. Effect of modified method of autonomic nervous system activity assessment on results of heart rate variability analysis. Acta Gymnica. 2013; 43 (2): 39-46.
7. Sociedade Brasileira de Imunizações (SBIm). Calendário de vacinação SBIm Criança (0-10 anos) - Recomendações SBIm 2022/2023. Brasil: SBIm; 2022 [access in 2022 Oct 31]. Available from:
https://sbim.org.br/images/calendarios/calend-sbim-crianca.pdf8. National Library of Medicine (NIH). National Center for Biotechnology Information. ClinVar - Genomic variation as it relates to human health; [VCV001519488.4]. [access in 2022 Dez 9]. Available from:
https://www.ncbi.nlm.nih.gov/clinvar/variation/1519488/9. Yıldırım İ, Topyıldız E, Bilgin RBG, Aykut A, Durmaz A, Edeer Karaca N,
et al. X-linked agammaglobulinemia: ınvestigation of clinical and laboratory findings, novel gene mutations and prevention of ınfective complications in long-term follow-up. Am J Clin Exp Immunol. 2021 Aug; 10 (2): 63-4.
10. Conley ME, Broids A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T,
et al. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005 Feb; 203 (1): 216-34.
11. Demirdag YY, Gupta S. Update on Infections in Primary Antibody Deficiencies. Front Immunol. 2021; 12: 634181.
12. Goudoris ES, Silva AMR, Ouricuri AL, Grumach AS, Condino-Neto A, Costa-Carvalho BT,
et al. II Brazilian Consensus on the use of human immunoglobulin in patients with primary immunodeficiencies. Einstein. 2017; 15 (1): 1-16.
13. Bundy V, Barbieri K, Keller M. Primary immunodeficiency: Overview of management. Up To-Date. 2021 [access in 2022 Jun 30]. Available from:
https://www.uptodate.com/contents/primary-immunodeficiency-overview-of-management14. Segundo GM, Condino-Neto A. Treatment of patients with immunodeficiency: Medication, gene therapy, and transplantation. J Pedriatr (Rio J. ). 2021; 97 (Supl. 1): S17-23.
Received on July 12, 2022
Final version presented on May 18, 2023
Approved on May 22, 2023
Associated Editor: Samir Kassar
Author´s contribution: Morais LJ: study design, data collection and analysis, manuscript writing and revision;
Silva BBM: data collection and analysis, manuscript writing;
Brito LAC, Lemos LAP, Lustosa MSL, Carneiro RRD and Aragão TG: data collection, manuscript writing;
Lima RCPC: study design, data analysis, manuscript revision;
Nóbrega VM: study design and supervision, data analysis, manuscript revision.
The authors approved the final version of the article and declare there is no conflict of interests.


 ; Beatriz Brasileiro de Macedo Silva2
; Beatriz Brasileiro de Macedo Silva2








 Ler em português
Ler em português