RESUMO
OBJETIVOS: apresentar um caso de Erro Inato da Imunidade (EII) como diagnóstico em potencial de pacientes pediátricos com infecções de repetição.
DESCRIÇÃO: paciente masculino, 13 anos, desde os oito meses de idade apresentou quadros repetidos de diarreias, sinusites, otites, abscessos e infecções do trato urinário; destacando-se a otite, sinusite e diarreia pela maior recorrência. Aos dez anos, quando apresentou mastoidite evoluindo para meningite, foi internado em um hospital terciário, onde foi realizada avaliação imunológica, a qual levou ao diagnóstico de Deficiência Predominantemente de Anticorpos (DPAs), tendo como suspeita a agamaglobulinemia ligada ao cromossomo X (ALX). Foi iniciado tratamento com administração de gamaglobulina endovenosa 400 mg/kg a cada quatro semanas, ocorrendo melhora significativa do quadro.
DISCUSSÃO: normalmente, o diagnóstico da ALX tende a ser feito nos primeiros três anos de vida. Neste relato, entretanto, embora as primeiras manifestações tenham iniciado aos oito meses de idade, ocorreu um atraso de dez anos até o início do tratamento. De fato, o diagnóstico de crianças e adultos com EII pode ser retardado se os profissionais de saúde não conseguirem encontrar a causa das infecções recorrentes. Destaca-se, portanto, a relevância de se considerar tais patologias na vigência de sinais de riscos, pois o diagnóstico precoce é fundamental para tratar e prevenir morbidades.
Palavras-chave:
Doenças da imunodeficiência primária, Imunidade, Infecções
ABSTRACT
OBJECTIVES: present a case of Inborn errors of immunity (IEI) as a potential diagnosis in pediatric patients with recurrent infections.
DESCRIPTION: male patient, 13 years old, since he was eight months old had recurrent diarrhea, sinusitis, otitis, abscesses and urinary tract infections. At the age of ten, he presented mastoiditis progressing to meningitis, he was admitted to a tertiary hospital, where an immunological evaluation was performed, which led to the diagnosis of Predominantly Antibody Deficiency (PAD), with suspected X-linked Agammaglobulinemia (XLA). Treatment was initiated with administration of intravenous gamma globulin 400 mg/kg every four weeks, with a significant improvement of the condition.
DISCUSSION: usually, the diagnosis of XLA tends to be made in the first three years of life. However, in this report, although the first manifestations started at eight months of age, there was a delay of ten years before starting the treatment. In fact, the diagnosis of children and adults with IEI can be delayed if healthcare professionals are unable to find the true cause of recurrent infections. Therefore, the relevance of considering such pathologies in the presence of risk signs is highlighted, as early diagnosis being essential in treating and preventing morbidities.
Keywords:
Primary immunodeficiency diseases, Immunity, Infections
IntroduçãoOs erros inatos da imunidade (EII) constituem um grupo com mais de 430 doenças que podem acarretar diversas manifestações no funcionamento do sistema imunológico. Dentre esse universo de patologias que constituem os EII, as deficiências predominantemente de anticorpos (DPAs) e a imunodeficiência combinada são as mais frequentes, sendo mais acometidos os indivíduos do gênero masculino e com consanguinidade na família. Essas patologias decorrem de defeitos genéticos, e sua expressão geralmente ocorre na infância, através de quadros de infecções de repetição, como otites, sinusites, estomatites e pneumonias. O diagnóstico precoce é fundamental para instituir o tratamento, assim como para definir qual o melhor manejo para o tipo de EII, prevenindo assim a morbidade que está atrelada ao quadro.
1-3A agamaglobulinemia ligada ao cromossomo X (ALX) está enquadrada na classificação dos EII em que há a predominância da deficiência de anticorpos. A ALX consiste na ausência ou diminuição de linfócitos B e de imunoglobulinas. Os indivíduos acometidos são suscetíveis a infecções bacterianas por agentes encapsulados e a enteroviroses. Em 85% dos casos, sua origem está atrelada a um erro no cromossomo X, gerando defeitos no gene da tirosina quinase Bruton (BTK), sendo por essa razão denominada agamaglobulinemia ligada ao X. O erro nesse gene acarreta níveis reduzidos ou ausência de níveis séricos de IgM, IgG, IgA, IgE e linfócitos B, assim como redução do tamanho de tonsilas, baço, adenoides e placas de Peyer. Isso ocorre devido ao BTK ser expresso em todas as linhagens de células hematopoiéticas, com exceção das células T e das células plasmáticas.
4No exame físico do paciente com ALX e alguns EII é comum o paciente apresentar as tonsilas e os linfonodos cervicais pequenos ou até ausentes.
1 Além disso, devido às otites de repetição, na otoscopia é comum observar lesões ou perfurações das membranas timpânicas. Em decorrência das infecções de vias aéreas superior e inferior, são frequentes as queixas de tosse crônica e secreção purulenta. Os estertores pulmonares crepitantes são recorrentes durante a ausculta pulmonar.
DescriçãoPaciente masculino, 13 anos, pardo, natural e procedente da cidade de Patos, na Paraíba. Aos oito meses, iniciou com um quadro de diarreia grave, com necessidade de internação, permanecendo desde então com recorrência de tal quadro. No primeiro ano de vida passou a apresentar sinusites que se tornaram recorrentes. Aos dois e três anos, desenvolveu abscesso de parede abdominal e abscesso de face com necessidade de internação. Aos sete anos, foi internado por hematúria e infecção do trato urinário. Após isso, foi acometido por otites, cujas culturas foram positivas para
Candidasp. e
Staphylococcus aureus, com um intervalo de cinco meses entre os quadros.
Aos dez anos, apresentou mastoidite evoluindo para meningite, foi internado no Hospital Universitário da capital do estado, sendo ampliada a avaliação imunológica, cujos resultados estão descritos na Tabela 1. Os exames mostraram diminuição de imunoglobulinas, linfócitos B ausentes, linfócitos T normais e células NK com níveis reduzidos, o que levou ao diagnóstico sindrômico de deficiência predominantemente de anticorpos (DPAs), sendo iniciado a administração de gamaglobulina endovenosa (GGEV) 400 mg/kg a cada quatro semanas. Como supracitado, os linfócitos B, células produtoras de anticorpos, encontram-se ausentes ou reduzidos na vigência dessa síndrome, enquanto os linfócitos T, em geral, não se alteram, pois fazem parte da imunidade celular.

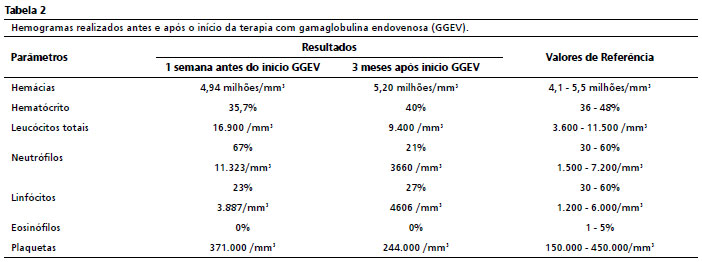
A partir disso, ocorreu melhora significativa do quadro, tanto clínica como laboratorialmente, conforme demonstrado nas Tabelas 1 e 2. Após iniciado o tratamento contínuo com a reposição mensal de GGEV, apresentou apenas um episódio de infecção do trato urinário, tendo a urocultura evidenciado
Escherichiacoli, utilizando-se ceftriaxona 30 mg/kg em tratamento domiciliar. Desde a primeira manifestação da imunodeficiência até o diagnóstico adequado, passaram-se dez anos. Durante esse tempo, o paciente perdeu um ano de estudo por faltas escolares, podendo também estar relacionada a perda auditiva mista de grau moderado na orelha direita, revelada pela avaliação audiológica.
Pensando em outras causas de imunodeficiências, foi solicitado o teste sorológico para o Vírus da Imunodeficiência Humana (HIV), cujo resultado foi "não reagente". Para confirmação etiológica da DPA, solicitou-se o teste genético que confirmou a principal hipótese de agamaglobulinemia ligada ao X, detectada por sequenciamento do exoma BTK em hemizigose.
Quanto aos antecedentes pessoais, a mãe refere ter realizado pré-natal, tendo o paciente nascido com 40 semanas de idade gestacional e pesado 3.435 kg. A queda do coto umbilical ocorreu no oitavo dia. Recebeu todas as vacinas do Programa Nacional de Imunização e não foi relatada reação adversa. O aleitamento materno exclusivo perdurou até os três meses de idade.
Quanto às doenças autoimunes, alérgicas e outras comorbidades, foi referido apenas vitiligo. Ao todo, foi hospitalizado sete vezes, com tempo máximo de 22 dias em uma hospitalização. Foram necessários cuidados intensivos durante a internação por meningite.
Não há consanguinidade na história familiar, porém contribuiu para o diagnóstico o falecimento do irmão de sua mãe aos 16 anos de idade por infecções recorrentes.
DiscussãoComo supracitado, as manifestações clínicas dos EII têm início durante a infância, embora, em alguns casos, possam ocorrer após a segunda ou terceira décadas de vida. O paciente relatado apresentou suas primeiras manifestações de forma bastante precoce, aos oito meses de idade. Em geral, o diagnóstico da agamablobulinemia congênita (AC) tende a ser feito mais comumente nos primeiros três anos de vida.
5 Neste relato, no entanto, o menino passou por um caminho debilitante de dez anos até a suspeita diagnóstica. De fato, o diagnóstico de crianças e adultos com EII pode ser retardado se os profissionais de saúde não conseguirem encontrar a verdadeira causa das infecções recorrentes. Isso já foi relatado em outros casos da literatura.
5,6O espectro de infecções nos EIIs são um dos principais fatores para a suspeição do diagnóstico na criança durante seus primeiros anos de vida, assim como também são um parâmetro de acompanhamento durante o tratamento.
4 Nesse sentido, o paciente do gênero masculino poderá ter a hipótese de diagnóstico para ALX considerada quando houver infecções recorrentes durante a primeira infância, histórico familiar patológico positivo para DPAs, ocorrência de internação hospitalar antes dos cinco anos de idade e reação vacinal, principalmente após vacina com vírus vivo atenuado. O paciente apresentou todas essas condições, com exceção das reações vacinais. A Sociedade Brasileira de Imunizações (SBIm) disponibiliza em seu arsenal para crianças de zero a cinco, as seguintes vacinas de vírus vivos atenuados: Poliomielite Oral, Rotavírus Monovalente, Rotavírus Pentavalente, Febre Amarela, Tríplice Viral (sarampo, caxumba e rubéola), Varicela e Dengue,
7 estas vacinas devem ser evitadas em pacientes com suspeita ou diagnóstico de ALX.
Os exames laboratoriais realizados para início da investigação da AC têm como objetivo confirmar a existência do defeito imunológico, sendo assim, devem conter hemograma, dosagem de imunoglobulinas (IgM, IgG, IgA e IgE) e titulação de anticorpos gerados em resposta às vacinas pertencentes ao quadro vacinal vigente. A segunda etapa de exames deve conter uma citometria de fluxo, a fim de fenotipar e contabilizar os linfócitos. No presente caso, após a avaliação imunológica, foi constatada a redução dos níveis séricos de imunoglobulinas e de linfócitos B, bem como a ausência de anticorpos contra rubéola e sarampo, além da presença de níveis séricos normais de linfócitos T. Além disso, a confirmação do diagnóstico pode ser feita por meio do teste genético que confirma variantes no gene BTK, ou, caso o teste genético não esteja disponível, através de um histórico familiar confirmado de ALX.
4 Como não foi possível confirmar o histórico familiar, solicitou-se o teste genético, que identificou, em hemizigose, no gene BTK, a variante ChrX:101.356.055 G>T, promovendo a substituição do aminoácido aspartato no códon 521 por glutamato (p.Asp521Glu). A partir dessa confirmação de agamaglobulinemia ligada ao X, classificou-se o paciente, através da classificação internacional de doenças (CID-10), como D80.0.
O aspartato na posição 521 é altamente conservado nas diversas espécies biológicas e programas computacionais de predição "
in sílico" de patogenicidade sugerem que sua substituição por glutamato seja potencialmente deletéria. Esta variante está ausente entre cerca de 181 mil cromossomos X em indivíduos de banco populacional e já foi previamente descrita na literatura médica associada ao quadro de agamaglobulinemia.
8,9 É válido ressaltar que o teste genético não é imprescindível para o início do tratamento na suspeita de um EII. No caso em questão, tal teste foi realizado apenas cerca de dois anos após o início da terapia.
A partir do diagnóstico sindrômico de DPAs, várias etiologias podem ser consideradas. Dentre os pacientes com células B circulantes abaixo de 2% do total de linfócitos e com níveis indetectáveis ou muito baixos de anticorpos, cerca de 85 a 90% apresentam ALX, devido a mutações no gene BTK, e 5% deles têm deficiência de cadeia pesada de imunoglobulina M, quadro cujas manifestações clínicas são mais graves em comparação à ALX. Os pacientes, nesse caso, apresentam um maior risco de sepse por pseudomonas, artrite, abscessos cutâneos, diarreia crônica e infecções enterovirais do sistema nervoso central (SNC).
10,11Já na imunodeficiência comum variável (ICV), há redução dos níveis séricos de pelo menos dois isotipos de imunoglobulinas, associada a quantidades normais ou reduzidas de células B. Geralmente, é diagnosticada entre a terceira e quinta década de vida, sendo que, quando iniciada na infância, costuma estar associada à otite média, déficit de crescimento e atraso no desenvolvimento.
10O tratamento para EII depende da doença diagnosticada e pode ser definitivo ou de suporte. As opções terapêuticas ainda estão em estudo e buscam prevenir e tratar infecções, estimular o sistema imunológico e tratar a causa da imunodeficiência. Diante da hipótese diagnóstica, a reposição da imunoglobulina humana é o principal método terapêutico em pacientes com EII em quase 75% desse grupo de doenças.
12 A administração pode ser feita por via intravenosa (GGEV) ou subcutânea (SCIg), entretanto em casos de urgência a GGEV é a via utilizada. A reposição endovenosa de imunoglobulina possui uma variação na dosagem entre 400 mg/kg/dose e 800 mg/kg/dose com administração realizada a cada 21 a 28 dias. A dose e o intervalo podem ser ajustados para diminuir a incidência de infecções. Já na reposição por via subcutânea preconiza-se iniciar a terapia usando 100 a 200 mg/kg/dose a cada sete dias.
12,13 O tratamento com imunoglobulina deve ser feito de forma contínua.
Após o início da terapia com imunoglobulinas, espera-se a redução das infecções graves, como sepse, meningite e celulite, todavia é comum que os pacientes continuem a apresentar infecções crônicas e/ou menos graves, como sinusites, infecções urinárias e otites.
2 Desde a suspeita diagnóstica de ALX, o paciente relatado iniciou a reposição de imunoglobulinas com administração endovenosa de IgG a cada quatro semanas, obtendo melhora considerável dos sintomas.
A profilaxia antibiótica é um outro método terapêutico e tem como objetivo diminuir os riscos de complicações, sendo específica para cada paciente. No caso em questão, não foi necessário fazer uso desse recurso até o momento.
Uma alternativa terapêutica para os EII é o transplante de células-tronco hematopoiéticas (TCTH), conhecido como transplante de medula óssea, que consiste na substituição de células alteradas por células-tronco hematopoéticas de doadores saudáveis. É uma opção de tratamento curativo para casos específicos de EII, contudo sua indicação deve ser individualizada e deve considerar os riscos de evolução futura da doença contra os riscos do transplante, como a doença do enxerto
versus hospedeiro. De forma geral, o TCTH não é indicado para pacientes com ALX.
14A terapia gênica, por fim, constitui uma possibilidade promissora de cura para portadores de EII, uma vez que corrige as alterações genéticas das células-tronco hematopoiéticas do indivíduo, restaurando as funções dos linfócitos B, sem haver necessidade de esperar por doadores compatíveis e com menor risco de rejeição.
14A partir da discussão do relato de caso em tela, pode-se afirmar que o diagnóstico dos EII representa, ainda, um desafio para os profissionais de saúde, pois muitos casos levam anos até receber o tratamento adequado. Recomenda-se, portanto, que pesquisas futuras estudem os principais fatores que impedem o diagnóstico precoce de tais doenças. Concomitantemente, é de fundamental importância que trabalhos de educação em saúde sejam realizados neste sentido, a fim de capacitar cada vez mais profissionais e, por conseguinte, evitar complicações decorrentes da doença, reduzir hospitalizações e gastos em saúde, bem como promover melhor qualidade de vida para os indivíduos acometidos.
Referências1. Rodrigues LS, Vernizzi CC, Ikuno CM, Menezes CT, Silva CM, Carvalho LR,
et al. Considerações sobre erros inatos da imunidade - um desafio diagnóstico na pediatria. In: FF Carvalho Junior. Alergia e imunologia: abordagens clínicas e prevenções. São Paulo: Científica Digital. 2021. p.55-77.
2. Ferreira JFS. Imunodeficiências primárias na infância - quando o pediatra deve suspeitar e como deve se conduzir? Rev Saúde Criança Adolesc. 2011; 3 (1): 58-62.
3. Pinto-Mariz F. Failure of immunological competence: when to suspect? J Pediatr (Rio J). 2021; 97 (Supl. 1): S34-8.
4. Vilela MM. Human Inborn Errors of Immunity (HIEI): predominatly antibody deficiencies (PADs): if you suspect it, you can detect it. J Pediatr (Rio J). 2021; 97 (Supl. 1): S67-74.
5. Estrella L, Foley ME, Cunningham-Rundles C. X-linked agammaglobulinemia in a 10-year-old child: A case study. J Am Acad Nurse Pract. 2007 Apr; 19 (4): 205-11.
6. Botek M, Krejčí J, Neuls F, Novotný J. Effect of modified method of autonomic nervous system activity assessment on results of heart rate variability analysis. Acta Gymnica. 2013; 43 (2): 39-46.
7. Sociedade Brasileira de Imunizações (SBIm). Calendário de vacinação SBIm Criança (0-10 anos) - Recomendações SBIm 2022/2023. Brasil: SBIm; 2022 [access in 2022 Oct 31]. Available from:
https://sbim.org.br/images/calendarios/calend-sbim-crianca.pdf8. National Library of Medicine (NIH). National Center for Biotechnology Information. ClinVar - Genomic variation as it relates to human health; [VCV001519488.4]. [access in 2022 Dez 9]. Available from:
https://www.ncbi.nlm.nih.gov/clinvar/variation/1519488/9. Yıldırım İ, Topyıldız E, Bilgin RBG, Aykut A, Durmaz A, Edeer Karaca N,
et al. X-linked agammaglobulinemia: ınvestigation of clinical and laboratory findings, novel gene mutations and prevention of ınfective complications in long-term follow-up. Am J Clin Exp Immunol. 2021 Aug; 10 (2): 63-4.
10. Conley ME, Broids A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T,
et al. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005 Feb; 203 (1): 216-34.
11. Demirdag YY, Gupta S. Update on Infections in Primary Antibody Deficiencies. Front Immunol. 2021; 12: 634181.
12. Goudoris ES, Silva AMR, Ouricuri AL, Grumach AS, Condino-Neto A, Costa-Carvalho BT,
et al. II Brazilian Consensus on the use of human immunoglobulin in patients with primary immunodeficiencies. Einstein. 2017; 15 (1): 1-16.
13. Bundy V, Barbieri K, Keller M. Primary immunodeficiency: Overview of management. Up To-Date. 2021 [access in 2022 Jun 30]. Available from:
https://www.uptodate.com/contents/primary-immunodeficiency-overview-of-management14. Segundo GM, Condino-Neto A. Treatment of patients with immunodeficiency: Medication, gene therapy, and transplantation. J Pedriatr (Rio J. ). 2021; 97 (Supl. 1): S17-23.
Recebido em 12 de Julho de 2022
Versão final apresentada em 18 de Maio de 2023
Aprovado em 22 de Maio de 2023
Editor Associado: Samir Kassar
Contribuição dos autores: Morais LJ: desenho do estudo, coleta e análise dos dados, escrita e revisão do manuscrito;
Silva BBM: coleta e análise dos dados, escrita do manuscrito;
Brito LAC, Lemos LAP, Lustosa MSL, Carneiro RRD e Aragão TG: coleta dos dados, escrita do manuscrito;
Lima RCPC: desenho do estudo, análise dos dados, revisão do manuscrito;
Nóbrega VM: desenho e supervisão do estudo, análise dos dados, revisão do manuscrito.
Os autores aprovaram a versão final do artigo e declaram não haver conflito de interesses.


 ; Beatriz Brasileiro de Macedo Silva2
; Beatriz Brasileiro de Macedo Silva2






 Ler em inglês
Ler em inglês